Electrochemistry
- An Adventure Into Nickel Cross Coupling Chemistry!
Here’s a behind the scenes look at one of our newest papers in JACS that highlights some nickel chemistry that we have developed! Phil has been interested for quite awhile now in transition metal cross-coupling methodology. A few ideas...
- A Tale Of Two Olefins
A prototype of the graphical abstract that Phil didn't want to submit to JACS.Today marks the day that another paper makes its way out of the hot little presses of the ACS Publication Office and into the world of chemistry. This time, it's a...
- Reactions
← Back to Organic Chemistry Problem-Solving Approach There are 4 types of questions: Given reactants + products → suggest reagents and conditions Given reactants + reagents → suggest products Given new reaction (Grignard, Killiani-Fishcer,...
- H2 Chemistry Syllabus (2008)
PHYSICAL CHEMISTRY 1. ATOMS, MOLECULES AND STOICHIOMETRY • Relative masses of atoms and molecules • The mole, the Avogadro constant • The calculation of empirical and molecular formulae • Reacting masses and volumes (of solutions and gases) 2....
- Introduction To Organic Chemistry : 12.5 Organic Reaction
TYPE OF CLEAVAGE : 2 type of bond cleavage:- Hemolytic cleavageHeterolytic cleavage Hemolytic cleavage The breaking of a single (two-electron) bond in which one electron remains on each of the atoms. Also known as free-radical reaction; homolysis. ...
Electrochemistry
Making C–C Bonds with the Simplicity of Making Amide Bonds
Despite all of the wonderful advances in organic synthesis, amide-bond formation is still the most widely used reaction among those making medicines. This truth has led some to declare their embarrassment and depression. Medicinal chemist Derek Lowe has a nice post reviewing this state of affairs. The authors of the J. Med. Chem. piece attribute the "over amide-ification" of medicinal chemistry to the following factors: "commercial availability of reagents, high chemoselectivity, and a pressure on delivery. "
What if one could piggyback on the popularity of amide bond formation – where people pay very little attention to things like the low atom-economy of coupling agents whose sole purpose is to expel water from an acid – and repurpose both the starting materials and reagents to instead accomplish a cross-coupling that would generate new C–C bonds with a loss of CO2? Such a reaction would probably find some use if the conditions and setup were nearly as simple as amide-bond formation and the process were chemoselective.
This brings us to our latest work in the field of nickel chemistry which was published today in Science. As a complement to our Ni-catalyzed aryl–alkyl cross-coupling paper from February, this paper features a general and practical sp3-sp3decarboxylative alkyl-alkyl cross-coupling.
This brings us to our latest work in the field of nickel chemistry which was published today in Science. As a complement to our Ni-catalyzed aryl–alkyl cross-coupling paper from February, this paper features a general and practical sp3-sp3decarboxylative alkyl-alkyl cross-coupling.
Thanks to 70+ years of amide bond formation, alkyl carboxylic acids are easily accessed from almost every commercial chemical supplier. They are everywhere. Considering their stability and general lack of toxicity, alkyl carboxylic acids should be ideal coupling partners in organic synthesis as the abundance of amides in Medicinal chemistry has taught us. In some ways they are really Nature's version of a boronic acid. Although the addition of a nucleophile or access to alternative oxidation states is known, utilizing simple alkyl carboxylic acids to perform decarboxylative cross-couplings is still rare.
In a nutshell, we believe that the redox-active esters used in these couplings, incidentally the same types of activated esters used to make amides (HOAt, HOBt, NHPI, etc.), will provide chemists a new tool to re-evaluate the retrosynthetic analysis of complex natural products and bioactive molecules. Here’s a behind the scenes look at the invention of this method.
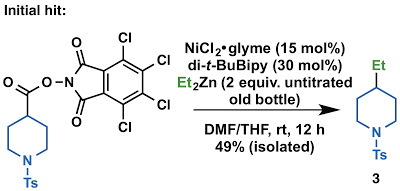
During our work on Ni-catalyzed aryl-alkyl coupling of aryl zinc reagents and alkyl carboxylic acids, we wondered if we could apply similar reaction conditions to forge sp3–sp3 bonds, historically a much more difficult problem than sp2–sp2 or sp2–sp3 cross coupling. The first bottle of diethylzinc I found was old and didn’t look too great, but the reaction worked even on the first attempt. Since we were interested in coupling more organometallic alkyl groups than just those that you can buy from a bottle (ZnEt2, ZnMe2, etc), we found that we could prepare dialkylzinc reagents from 2 equivalents of alkyl Grignard reagent or alkyl lithium reagent mixed with 1 equivalent of a ZnCl2 solution. This solution could directly be used for the cross-coupling reaction (we didn’t need to remove the Mg or Li salts formed from the transmetallation). The procedure is also simple to do (even one of our undergraduate students managed to prepare the dialkylzinc reagents and perform the decarboxylative coupling in his first trial).
With this simple and practical protocol, I found that almost every possible dialkylzinc reagent (besides dibenzylzinc) worked great in the cross-coupling reaction. Moreover, this reaction was found to be general for primary and other secondary redox-active esters. After I showed the preliminary table to Phil, he looked at it for a few seconds and asked me: “As a molecule maker, don't you think that this methodology could change people’s thoughts about retrosynthetic analysis?” After thinking about this and running these reactions for the past few months, I am totally convinced that this methodology could provide the community a C–C bond forming reaction analogous in simplicity, utility, and practicality to classic amide bond formation.
With Jacob and Pep’s great help, we finished more than 40 total substrates in two weeks. Some of the more interesting examples include the use of feedstock acids like adipic acid, tartaric acid, and 2-fluoropropionic acid. We also sought out more complex coupling partners, like pharmaceuticals (such as atorvastatin, cetirizine) and complex natural products (cholic acid and biotin). Proximity to the Cravatt lab gave us lots of fatty acids to play with as well. Unlike alkyl halides, you can't throw a stone in a typical chemistry lab without hitting a carboxylic acid. Also a shout-out to Brad Maxwell of BMS for running the radio labeled example that demonstrates that one can exchange –CO2H for any radio labeled carbon source you like.
| Some selected examples from ca. 60 reported. |
Another defining feature of this work is the ability to couple α-oxy acids readily and efficiently. While most α-oxy-halogen substituted compounds are unstable, and thereby unusable in cross-coupling chemistry, α-oxy acids could smoothly convert to different types of ethers, some of which are hard to synthesize through traditional Williamson ether synthesis (which is incidentally another one of the most used reactions in Medicinal chemistry). Although α-oxy acids work well in this chemistry, one current limitation (that we are working hard to solve) is that it is difficult to couple benzylic acids as well as benzylic zinc reagents in this chemistry currently.
| An alternative to the Williamson ether synthesis... |
As the Baran lab peptide synthesis aficionado, Lara came on board to test the alkyl-alkyl cross-coupling reaction in one final chemoselectivity challenge: the direct modification of native peptides on the solid-phase. After all, if one has a reaction that is supposedly as easy as amide-bond formation, it better work on solid-phase. She prepared a handful of model peptides on Rink amide resin, each bearing a free side-chain (aspartic acid or glutamic acid) or a free α-carboxylic acid. The acids were activated on-resin as redox-active esters and then subjected directly to solid-phase nickel couplings with various diaklylzinc reagents. Before cleavage of the peptides from the solid support, all excess reagents were simply washed away, identical to the way excess peptide coupling reagents are washed away after amide bond formation on the solid-phase. In this way, we were able to repurpose age-old solid-phase synthesis techniques for late-stage, nickel-catalyzed C-C bond formation. Accessing the types of compounds shown below would otherwise require the synthesis of each unnatural amino acid (usually a multi-step synthesis!) and its incorporation into a new resin-bound peptide. We think the ease with which we can rapidly install valuable, non-native functional groups (e.g. alkenes for bioconjugation or peptide stapling) onto completely native peptides is one major advantage of the solid-phase method. Given the potential for late-stage peptide diversification, we also hope that this technique will find applications in both academia and industry for the rapid synthesis of peptide drug analogues. Finally it's interesting to contemplate the use of this method for iterative organic synthesis without having to invent any new equipment or building blocks...
| If it's as easy as amide-bond formation it better work on a solid phase... |
After we demonstrated the transformation on various primary and secondary carboxylic acids, including resin-bound peptides, we were curious to find if we could extend this work to tertiary carboxylic acids. Due to the difficulties associated with cross-coupling of tertiary electrophiles, we considered this a Holy Grail of cross-coupling chemistry. Fortunately, Pep and Shuhei found that 6,6’-dimethylpyridine was the sweet ligand for this transformation. With a few other changes to the reaction conditions, tertiary bridgehead acids could be used to obtain the desired products in decent yield; however, no product was observed for other tertiary radicals. Although we couldn’t use tertiary acids for a direct cross-coupling reaction with alkylzinc reagents, Chao came up with a brilliant idea to use the stability of the tertiary radical to our advantage. The hypothesis that we had is that in the presence of a radical acceptor, such as an alpha-beta unsaturated ester, we might be able to trap the tertiary radical and then subsequently do a coupling with an organozinc reagent, such as PhZnCl. Well, as it would turn out, this totally works in good yield! This three-component coupling works well for both normal, unstabilized tertiary radicals as well as α-heteroatom stabilized radicals.
| A 3-component coupling that makes quaternary centers with great ease. |
Similar to our previous Ni coupling reaction, this one is also easy to run; it doesn’t require rigorous drying of glassware, no fancy equipment is required, and you get to see dramatic color changes (from deep red to deep green)! Even for our peptide synthesis escapades, specialized equipment for solid-phase synthesis wasn’t required to perform any of the reactions. All peptide synthesis was done manually (though occasionally involving some creative use of the rotovap as a heating and stirring apparatus), and all you need to get started is a solid-phase reaction vessel and a suitable resin (e.g. Rink amide).
On a less serious note, we were curious if we could take carboxylic acids that you find at the grocery store or hardware store, such as biotin (Vitamin H), cetirizine (Zyrtec), isobutyric acid (in Vanilla), and 2,4-Dichlorophenoxyacetic acid (2,4-D, an herbicide), and use these products directly in the cross-coupling reaction.
 |
| Acids are literally everywhere. Some common products containing alkyl carboxylic acids. As shown in this video, even an office jockey like Phil can get back in the hood and successfully make both an amide and a C-C bond. Click here to see which of the commercial acids in the picture above he chose to do the coupling on. The results are surprising... Finally, I wanted to point out that the SI for this paper (ca. 460 pages) has been carefully crafted with pictures of everything, a FAQ section, extensive mechanistic discussion/experiments, and even a troubleshooting section. Phil's request to us was to formulate a document that would answer as many questions as possible and allow for extreme ease in reproducibility without any ambiguity and we tried our best to do that. Let us know if you have any questions and thanks for reading. |
-Tian
- An Adventure Into Nickel Cross Coupling Chemistry!
Here’s a behind the scenes look at one of our newest papers in JACS that highlights some nickel chemistry that we have developed! Phil has been interested for quite awhile now in transition metal cross-coupling methodology. A few ideas...
- A Tale Of Two Olefins
A prototype of the graphical abstract that Phil didn't want to submit to JACS.Today marks the day that another paper makes its way out of the hot little presses of the ACS Publication Office and into the world of chemistry. This time, it's a...
- Reactions
← Back to Organic Chemistry Problem-Solving Approach There are 4 types of questions: Given reactants + products → suggest reagents and conditions Given reactants + reagents → suggest products Given new reaction (Grignard, Killiani-Fishcer,...
- H2 Chemistry Syllabus (2008)
PHYSICAL CHEMISTRY 1. ATOMS, MOLECULES AND STOICHIOMETRY • Relative masses of atoms and molecules • The mole, the Avogadro constant • The calculation of empirical and molecular formulae • Reacting masses and volumes (of solutions and gases) 2....
- Introduction To Organic Chemistry : 12.5 Organic Reaction
TYPE OF CLEAVAGE : 2 type of bond cleavage:- Hemolytic cleavageHeterolytic cleavage Hemolytic cleavage The breaking of a single (two-electron) bond in which one electron remains on each of the atoms. Also known as free-radical reaction; homolysis. ...